Describe the general properties of the complexity of mRNA populations? Why is it important to consider these properties when planning a gene expression experiment
A population of mRNA extracted from a cell contains not just a lots of different RNA species, but also each with different level of expression. Most of the highest expressed genes are housekeeping genes, which are always expressed in high level to maintain cell activity. Other highest expressed genes are the characteristic of the cell, for example, mitochondrial genes are most expressed in metabolically active cells, or developmental pluripotency associated 5A (Dppa5a) in pluripotent cell. Meanwhile, most of the genes have low expression levels, because they are not expressed.
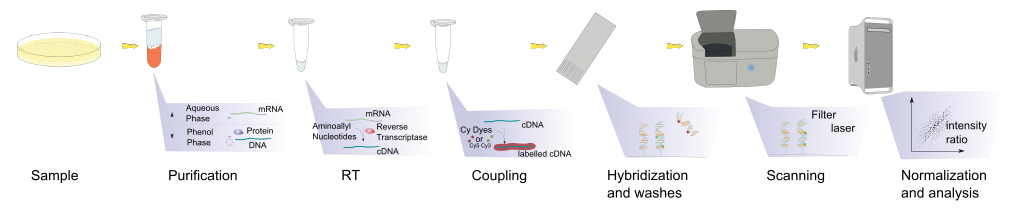
Hybridisation allows RNA expression levels to be measured
So, in an mRNA population, the most abundant molecules will be dominated by several genes. The problem with this is, sometimes the genes that we find or interested in, are expressed in low levels. In gene expression experiment, what we want to do is comparing mRNA populations between samples, and see which gene are expressed differentially. So, we will probably see similar mRNA abundance distribution even in different cells. And with this, we can also see if there are contaminations (because we expect similar trends).
When the change happens in a lower abundance level (compared to the housekeeping genes for example), it will be hard to detect those. This is true especially when dealing with microarray experiment.
Therefore, we must consider giving the right amount of probe in microarray experiment, because giving more probe to microarray increases the intensity level, but also increses the background intensity.
This happens because the basic of microarray is hybridization. So, a probe-set will complement with its target (specific binding). But, even if the array are able to separate those species through its complementary with certain mRNA, non-specific binding will occur randomly. And this non-specific binding will statistically, caused by the most abundant number of mRNA species in the sample, which contribute to overall background. Increasing probe will also increase the chance of non-specific binding. Therefore it limits the detection level of microarray experiment to measure lower level genes.
You cannot put more probe to detect lower expression in microarray, it just increases background. When you want to detect something rare, we can make more probe of that at lower abundance, but when we do that we are also labelling all the other guys which is not interesting, and all give background.
So, one thing to consider is that microarray relies on the balance between specific binding and non-specific binding. Because microarray measure specific signal against a background of non-specific hybridisation. DNA hybridisation kinetics limit the amount of signal that can be obtained from weakly expressed genes. Labelling more probe label does not generally increases detection because common trancsripts always obtain most of the label.
But nevertheless, sometimes we can see a high number of Trancription Factor in an mRNA population, which connected to a genes.
Outline how PMA (Present, Marginal, Absent) calls are generated from Affymetrix!
To probe genes, oligonucleotides
of length 25 bp are used (2). Typically, a mRNA
molecule of interest (usually related to a gene) is represented
by a probe set composed of 11±20 probe pairs of these
oligonucleotides. Each probe pair is composed of a perfect
match (PM) probe, a section of the mRNA molecule of
interest, and a mismatch (MM) probe that is created by
changing the middle (13th) base of the PM with the intention
of measuring non-speci®c binding.
In Affymetrix Array, each probesets refer to one specific mRNA or genes. These probesets are a set of 25-ish oligonucleotide which complement to specific mRNAs or genes or predicter exons. The probeset pair composed of two types, which are the Perfect Match probe (PM), and mismatch probe (MM). There is only one single nucleotide differences in the MM. The idea is that the PM will only pair with specific-binding, and the MM will pair with non-specific binding. Therefore, the hybridisation intensity of the mismatch probe is subracted from the perfect match probe to get the correct intensity of specific hybridisation.
In Affymetrix array, the resulting hybridisation scanned into an image file (DAT) which then summarized into a CEL file. This CEL file is the average reads of all the probesets. Then, summary algorithm was generated to create two types of information, one is the number of the signal (the intensity) and the other is the probability of confidence that the gene is expressed or a flag.
What does each call tell you about the expression level of a given gene?
The PMA flag indicates confidence that a particular measurement is from the signal (match) and not from the background (mismatch). This is done by comparing the signal between the PM and MM. Basically it just say how many PM value is higher (brighter) than the corresponding MM value. If tou pick a random intensities, it should result in 50%. Then, we use Fisher's rank sign test to generate p-value. And we can use this significance value from PMA calls as threshold of confidence whether it is:
- P = present, high value, real measurement of expression
- M = marginal, intermediate value, probably expressed
- A = absent, small value, no evidence that signal is different from the background
So, standard image analysis results in image value (signal intensity) and the confidence that the value is from the perfect match signal.
Even so, designing mismatches are not so clever because for example, humans have a lot of Single Nucleotide Polymorphisms. The idea of match/mismatch probes could end up not working because the mismatch probe almost hybridises as well as the perfect match probe. Resulting in subtracting both from each other generate no signal, thus leads to false conclusion. Hence, today we use statistical approaches like RMA (quantile normalisation) which only utilise the PM probe.
How could you use a PMA call alongside fold change filtering of gene to identify deferentially expressed genes?
To de®ne a measure of expression representing the amount
of the corresponding mRNA species it is necessary to
summarize probe intensities for each probe set. Several
Fold change takes the ratio of a gene's average expression levels under two conditions. It is usually calculated as the difference on the log2 scale.
Let
x ij be the log-transformed expression measurement of the
i th gene on the
j th array under the control (
i = 1,
⋯,
n and
j = 1,
⋯,
m0), and
y ik be the log-transformed expression measurement of the
i th gene on the
k th array under the treatment (
k = 1,
⋯m1). We define

and

.
Fold change is computed by
As for the traditional
t test, it is usually calculated on the
log2 scale to adjust for the skewness in the original gene expression measurements. The
t statistic is then computed by
where

is the pooled variance of
x ij and
y ik . Comparing (1) and (2), it is obvious that fold change and
t statistic are based on two contradicting assumptions.
The underlying assumption of fold change is that all genes share a common variance (on the
log2 scale), which is implied by the omission of the variance component in (1). On the other hand, the inclusion of
in (2) suggests that
t test assumes gene-specific variances.
In order for a gene to be flagged as DE, the double filtering procedure would require the gene to have extreme test scores under the common variance assumption as well as under the gene-specific variance assumption. It is analogous to using the intersection of the rejection regions defined by fold change and t statistic.
Assuming a common variance for all the genes apparently is an oversimplification. The assumption of gene-specific variances, however, leads to unstable estimates due to limited replicates from each gene. A more realistic assumption might lie in between the two extremes, i.e., modeling gene variances by a mixture of two components, one being a point mass at the common variance, another being a continuous distribution for the gene-specific variances. Under this mixture variance assumption, a DE gene could have a large fold change or a large
t statistic, but not necessarily both. Taking intersection of the rejection regions flagged by fold change and
t statistic, as is adopted by the double filtering procedure, might not be the best strategy under the mixture variance assumption.
MAS5 vs RMA
A significant challenge with Affymetrix expression data is to provide an algorithm that combines the signals from the multiple Perfect-Match (PM) and Mismatch (MM) probes that target each transcript into a single value that sensitively and accurately represents its concentration.
MAS5.0 does this by calculating a robust average of the (logged) PM-MM values [1]; increased variation is observed at low signal strengths and is at least in part due to the extra noise generated by subtracting the MM values from their PM partners [2].
- MAS5 normalises each array independently and sequentially; RMA as the name suggests (robust multi-array) uses a multi-chip model
- MAS5 uses data from mismatch probes to calculate a "robust average", based on subtracting mismatch probe value from match probe value
- RMA does not use the mismatch probes, because their intensities are often higher than the match probes, making them unreliable as indicators of non-specific binding
- RMA values are in log2 units, MAS5 are not (so values are not directly comparable)
Detection calls
In addition to expression summaries, the Affymetrix software also generates a p-score that assesses the reliability of each expression level. This is produced by using a signed rank test to consider the significance of the difference between the PM and MM values for each probeset [
9]. Informally, MAS 5.0 can be seen to return two values, the first, an estimate of transcript concentration, and the second, a measure of how much the software 'believes' the first. Of potential confusion is the fact that this value is referred to as the 'detection p-value', and is subsequently used to generate a 'detection call', which flags the transcript as 'Present', 'Marginal' or 'Absent' (P/M/A)